Lipid metabolism plays a critical role in maintaining cellular function, energy balance, and tissue homeostasis. The intricate network of lipids in the body contributes to numerous biological processes, including membrane structure, signaling, and energy storage. However, when lipid metabolism is dysregulated, it can lead to a variety of diseases, ranging from metabolic disorders to cardiovascular diseases and even certain neurodegenerative conditions. As our understanding of lipid biology deepens, new therapeutic approaches targeting lipid metabolism are emerging as promising strategies for treating a wide range of diseases. One such approach is the regulation of lipid peroxidation, a process that can have both beneficial and detrimental effects on health.
The Role of Lipids in Health and Disease
Lipids are essential molecules that serve as key components of cellular membranes, energy storage molecules, and signaling molecules. They are broadly classified into categories such as phospholipids, glycolipids, cholesterol, and fatty acids, each of which has distinct roles in cellular function. For instance, cholesterol is crucial for membrane fluidity and the formation of lipid rafts that facilitate cellular signaling. Meanwhile, fatty acids, especially unsaturated fatty acids, are involved in energy production and signaling, influencing inflammation and immune responses.
However, when lipid metabolism is disrupted, it can result in several diseases. Dyslipidemia, characterized by abnormal levels of lipids (such as cholesterol and triglycerides), is a major risk factor for cardiovascular diseases like atherosclerosis and heart attack. Similarly, obesity and type 2 diabetes are closely linked to impaired lipid metabolism, leading to insulin resistance and chronic inflammation. In recent years, the role of lipids in diseases like cancer and neurodegenerative disorders such as Alzheimer’s disease has also gained increasing attention.
Lipid Peroxidation: A Double-Edged Sword
One of the most critical aspects of lipid metabolism is the process of lipid peroxidation. Lipid peroxidation refers to the oxidative degradation of lipids, primarily polyunsaturated fatty acids. This process occurs when free radicals, such as reactive oxygen species (ROS), attack the double bonds in lipids, resulting in the formation of reactive metabolites known as lipid peroxides. These lipid peroxides can further decompose into a variety of harmful byproducts, including malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE), which can damage cellular structures and contribute to disease development.
In small amounts, lipid peroxidation plays an essential role in signaling pathways, including those involved in immune response and cellular communication. However, when excessive, it leads to oxidative stress, a condition that is implicated in a range of diseases. Chronic oxidative stress from lipid peroxidation can damage DNA, proteins, and cell membranes, contributing to the pathogenesis of diseases like cancer, cardiovascular diseases, and neurodegeneration.
Moreover, lipid peroxidation is closely linked to inflammation, as it can activate inflammatory pathways that exacerbate disease progression. The NF-κB and NLRP3 inflammasome signaling pathways, which are involved in the immune response, can be triggered by lipid peroxides, amplifying the inflammatory environment.
Targeting Lipid Peroxidation for Therapeutic Intervention
Given the central role of lipid peroxidation in disease development, strategies to modulate this process have become a key focus of therapeutic research. One promising approach is the use of small molecules or inhibitors that regulate lipid metabolism and prevent excessive lipid peroxidation. Liproxstatin-1, a selective inhibitor of lipid peroxidation, is one such compound that has garnered significant attention for its potential to combat diseases associated with oxidative stress and lipid damage.
By specifically targeting the lipid peroxidation pathway, Liproxstatin-1 prevents the accumulation of harmful lipid peroxides and mitigates the oxidative damage that can lead to cellular dysfunction and tissue damage. Research suggests that Liproxstatin-1 may have applications in several diseases, especially those where lipid peroxidation plays a pivotal role in disease pathogenesis.
Liproxstatin-1 in Neurodegenerative Diseases
One of the most promising applications of Liproxstatin-1 lies in the treatment of neurodegenerative diseases, where oxidative stress and lipid peroxidation are believed to play a significant role in disease progression. In conditions like Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS), neurons are particularly vulnerable to damage from reactive oxygen species and lipid peroxidation. This damage contributes to neuroinflammation, neuronal death, and the accumulation of toxic protein aggregates, such as amyloid plaques in Alzheimer’s or Lewy bodies in Parkinson’s.
By inhibiting lipid peroxidation, Liproxstatin-1 may help protect neurons from oxidative damage and reduce the progression of neurodegenerative conditions. Studies have shown that Liproxstatin-1 can preserve neuronal function and mitigate cognitive decline in preclinical models of Alzheimer’s disease, suggesting that targeting lipid peroxidation could be a novel therapeutic approach for slowing the progression of these devastating diseases.
Cardiovascular Diseases and Lipid Peroxidation
Another area where Liproxstatin-1 shows promise is in the treatment of cardiovascular diseases, particularly atherosclerosis. Atherosclerosis is characterized by the buildup of lipid-rich plaques in the arteries, which is driven in part by oxidative stress and lipid peroxidation. The accumulation of lipid peroxides in the arterial wall can lead to endothelial dysfunction, inflammation, and plaque formation, ultimately resulting in arterial blockages and heart attacks.
By reducing lipid peroxidation, Liproxstatin-1 has the potential to limit plaque formation and improve vascular health. In preclinical models, it has been shown to decrease oxidative stress and reduce inflammatory markers, making it a promising candidate for preventing or treating atherosclerosis and other cardiovascular conditions.
Cancer and Lipid Peroxidation
Lipid peroxidation also plays a significant role in cancer development. Excessive oxidative damage from lipid peroxides can lead to mutations in DNA and the activation of pro-tumorigenic signaling pathways. Interestingly, while lipid peroxidation can drive tumor progression, some cancer cells are addicted to oxidative stress, relying on it for growth and survival. In these cells, inhibiting lipid peroxidation with molecules like Liproxstatin-1 may impair their ability to thrive and spread, potentially sensitizing tumors to other therapeutic interventions.
Research is ongoing to understand how lipid peroxidation inhibitors can be integrated into cancer therapies, especially in cancers where oxidative stress plays a critical role in disease progression. Liproxstatin-1’s ability to selectively inhibit lipid peroxidation could be used in combination with conventional treatments like chemotherapy or radiation therapy to enhance their efficacy and reduce side effects.
Future Directions and Challenges
Despite the promising results from preclinical studies, there are still challenges to overcome before Liproxstatin-1 and similar compounds can be widely used in clinical settings. One key challenge is ensuring the selective inhibition of lipid peroxidation without disrupting essential cellular functions that rely on moderate oxidative signaling. Another hurdle is understanding the optimal dosage and delivery methods for these compounds to maximize therapeutic benefits while minimizing potential side effects.
Furthermore, as with any emerging drug candidate, the safety and long-term effects of Liproxstatin-1 need thorough evaluation through clinical trials before it can be adopted as a standard treatment. However, the growing body of evidence supporting the therapeutic potential of lipid peroxidation inhibitors offers hope for novel treatment options for a variety of diseases linked to oxidative stress.
Conclusion
Lipid metabolism is central to maintaining cellular and tissue homeostasis, but when it goes awry, it can lead to a range of diseases, including cardiovascular diseases, neurodegenerative conditions, and cancer. The regulation of lipid peroxidation represents an exciting therapeutic avenue, with compounds like Liproxstatin-1 showing promise in preclinical studies. By selectively inhibiting lipid peroxidation, Liproxstatin-1 has the potential to offer new treatments for diseases characterized by oxidative stress and lipid damage. As research in this area progresses, targeting lipid metabolism could pave the way for more effective and precise treatments for a wide range of diseases.
Liproxstatin-1 Attenuates SAP-Induced Acute Kidney Injury by Inhibiting Ferroptosis
Abstract
Background Acute kidney injury AKI is a frequent complication of severe acute pancreatitis SAP. Ferroptosis is involved in a range of diseases. However, the role of ferroptosis in SAP-induced AKI has yet to be elucidated.
Aims We aimed to investigate whether ferroptosis is induced in the kidney after SAP and whether inhibition of ferroptosis ameliorates AKI in a rat model of SAP.
Methods Sodium taurocholate five percent was retrogradely perfused into the biliopancreatic duct to establish a model of SAP with AKI in rats. The levels of serum amylase, lipase, tumor necrosis factor TNF-alpha, interleukin IL-6, creatinine Cr and blood urea nitrogen BUN in rats were measured. We also determined the biochemical and morphological changes associated with ferroptosis in renal tissue, including iron accumulation, lipid peroxidation assays, and mitochondrial shrinkage. H&E staining was used to assess pancreatic and renal histological changes. Western blot analysis, RT-PCR, and immunofluorescence staining were performed to analyze the expression of ferroptosis-related proteins and genes.
Results SAP-induced AKI was followed by iron accumulation, increased lipid peroxidation, and upregulation of ferroptosis-related proteins and genes. Twenty-four hours after SAP, TEM confirmed the presence of typical shrunken mitochondria. Furthermore, treatment with liproxstatin-1 lowered the levels of serum amylase, TNF-alpha, IL-6, Cr and BUN, decreased kidney lipid peroxidation and alleviated pancreatic and renal histopathology injury in SAP rats.
Conclusion Our findings are the first to demonstrate the involvement of ferroptosis in SAP-associated renal damage and present ferroptosis as a therapeutic target for effective treatment of SAP-induced AKI.
Keywords Ferroptosis, Acute kidney injury, Severe acute pancreatitis, Liproxstatin-1
Introduction
Severe acute pancreatitis SAP, an acute inflammatory condition of the pancreas, is a potentially lethal disease with considerable morbidity and mortality. It is often accompanied by systemic inflammatory response syndrome SIRS, sepsis, and even multiple organ dysfunction syndrome MODS. Acute kidney injury AKI is one of the most common complications of SAP, along with acute respiratory distress syndrome, acute liver injury and intestinal mucosal damage, and increases disease mortality. Acute renal failure is defined as a severe form of AKI that leads to drastic increases in mortality associated with SAP. However, the underlying mechanisms of AKI in patients with SAP are not clear. Therefore, a better understanding of AKI in the context of SAP may aid in the identification of novel therapeutic targets.
The mechanism by which SAP results in AKI is complex. It has been suggested that SAP-induced AKI is mainly related to SIRS, which involves various cytokines and inflammatory mediators, such as tumor necrosis factor TNF-alpha, high-mobility group box protein 1 HMGB1, interleukin IL-1beta, IL-6, IL-10, and nuclear factor kappa B NF-kappa B. Production of inflammatory mediators, including complement activation products, and accumulation of reactive oxygen species ROS, which have been demonstrated to play critical roles, are fundamental causes of SAP-induced AKI. ROS can directly damage the structure and function of the kidney and can also induce and amplify the inflammatory response through oxidative stress, aggravate renal injury, and induce apoptosis. Previous studies have revealed that other types of cell death may be involved in AKI in addition to apoptosis. However, whether inflammatory mediators and oxidative stress induced ferroptosis of renal tubular epithelial remains unknown.
Ferroptosis, a recently recognized form of regulated cell death RCD that involves iron accumulation and lipid peroxidation, is remarkably distinguishable from other forms of RCD, including necroptosis, apoptosis, and unregulated necrosis, in terms of its biochemistry, morphology, and genetics. Ferroptosis has been declared to be involved in many diseases, such as neurological diseases, myocardial infarction, ischemia/reperfusion I/R injury IRI, and cancer. ROS accumulation has also been studied as a crucial factor in ferroptosis induction. Glutathione peroxidase 4 GPX4, a lipid repair enzyme, is the central regulator of ferroptosis. In addition, acyl-CoA synthetase long-chain family member 4 ACSL4 was recently found to facilitate the esterification of arachidonoyl AA and adrenoyl into phosphatidylethanolamine PE in a process closely related to ferroptosis. ACSL4 is the key enzyme to regulate lipid composition in ferroptosis, and the abnormal expression or dysfunction of ferritin heavy chain 1 FTH1 will lead to the increase of intracellular iron concentration, which will contribute to the execution of ferroptosis. Moreover, this form of cell death, which is regulated by a distinct set of genes including ACSL4, IREB2, GPX4, and SLC7A11, can be distinguished morphologically by the existence of shrunken mitochondria and can be inhibited specifically by ferrostatin-1 and liproxstatin-1 Lip-1. Recent studies have indicated that ferroptosis plays an important role in IRI-induced acute tubular necrosis ATN and rhabdomyolysis-associated renal damage. However, the precise mechanisms underlying ferroptosis in AKI with SAP and whether the inhibition of ferroptosis might protect SAP-associated renal injury remain unknown.
In the present study, we sought to confirm the occurrence of ferroptosis in the kidney after SAP by investigating iron and lipid peroxidation product accumulation and by examining ultrastructural changes in the mitochondria. Furthermore, we tested the efficacy of Lip-1, a specific inhibitor of ferroptosis, as a therapeutic agent for renal injury in rats. This study will advance our understanding of cell death pathways associated with SAP-induced AKI and will provide a new therapeutic approach for SAP-associated renal injury.
Materials and Methods
Animals and Model The animal procedures used in this study were performed according to the National Institutes of Health guidelines for laboratory animals and were approved by the Animal Care and Experiment Committee of Qingdao University Qingdao, China. Seventy-two adult male Sprague–Dawley rats age, 8–10 weeks; weight, 200–250 g were obtained from the animal center of Qingdao University Qingdao, China. The rats were randomly assigned to three groups: the sham control group, the vehicle-treated SAP group and the Lip-1-treated SAP SAP + Lip-1 group. The rats in the three groups were then divided into 6, 12 and 24 h time-point subgroups each subgroup contained 8 rats.
The rats were fasted for 12 h prior to the experiments. After anesthesia with 3 percent sodium pentobarbital 20 mg/kg, the SAP model was induced by standard pressure-controlled infusion of a freshly prepared 5 percent sodium taurocholate Sigma solution 1 ml/kg into the biliopancreatic duct. In the sham control group, the duct was infused with an equal quantity of sterile saline. In the SAP + Lip-1 group, Lip-1 Selleck, a ferroptosis inhibitor, was administered i.p. at a concentration of 10 mg/kg 1 h before the establishment of the SAP model, following previous study protocols. The sham group and the SAP model group were injected with equal volumes of 2 percent dimethyl sulfoxide DMSO.
Sample Collection The rats from each group were anesthetized again at designated time points after the induction of pancreatitis. Blood samples were collected through postcava puncture and then centrifuged to obtain the serum, which was stored at minus 20 degrees Celsius until assay. Pancreas and kidney tissues were harvested; a subset of tissues was immediately fixed in 4 percent paraformaldehyde for tissue section preparation, while the rest of the tissues were stored at minus 80 degrees Celsius for later analysis.
Serum Assays Serum amylase AMY and lipase LPS levels in the blood samples were determined with an automatic biochemistry analyzer Olympus Corporation, Tokyo, Japan. The concentrations of TNF-alpha, IL-6, creatinine Cr, and blood urea nitrogen BUN were measured using standard diagnostic kits Jiancheng Biotech, Nanjing, China following the manufacturer’s instructions.
Iron Measurements Fresh kidney tissues were immediately homogenized with phosphate-buffered saline PBS. The supernatant was collected after centrifugation. The iron levels were detected with an Iron Assay Kit Abcam according to the manufacturer’s instructions.
Lipid Peroxidation Assays To determine the lipid peroxidation levels at different time points after SAP, we collected renal tissues from the three groups and analyzed the malondialdehyde MDA and glutathione GSH levels and GPX4 activity by using commercially available kits Jiancheng Biotech.
Transmission Electron Microscopy TEM The renal tissues were washed with precooled PBS pH 7.4 and then postfixed in phosphate-buffered glutaraldehyde 2.5 percent and osmium tetroxide 1 percent. The kidney samples were then cut and stained en bloc with 2 percent uranyl acetate UA, dehydrated in a graded ethanol series, and embedded in an epoxy resin. Then, the sections 70–90 nm were stained with UA and lead citrate. Ultrastructural images were captured with a transmission electron microscope Hitachi HT7700, Tokyo, Japan.
Tissue Histology and Immunofluorescence For histological analysis, rat pancreas and kidney tissues were fixed in 4 percent formalin and then paraffin-embedded. The specimens were cut in 4-micrometer-thick sections for staining with hematoxylin and eosin H&E. The sections were then examined with a light microscope. Pathological scores were blindly evaluated by two independent pathologists according to previously established scoring criteria.
The expression of ACSL4 and GPX4 was determined by immunofluorescence analysis. Cryostat sections 4 micrometers thick were fixed with 4 percent paraformaldehyde fixative at room temperature for 20 min and then washed in PBS three times for 3 min. The sections were then incubated with a blocking solution normal goat serum at room temperature for 20 min. Next, the sections were incubated with rabbit anti-ACSL4 Abcam or rabbit anti-GPx4 Abcam primary antibodies overnight at 4 degrees Celsius. After the sections were washed three times for 3 min each with PBS, they were incubated with secondary antibodies for another hour at room temperature. The sections were then rewashed with PBS, coverslip-mounted, and observed and photographed under a fluorescence microscope Olympus, Tokyo, Japan.
Western Blot Analysis The levels of ACSL4, GPX4, and FTH1 in kidney tissues were detected by western blot analysis. Total proteins were extracted from the rat kidneys and quantified by using a BCA Protein Assay kit TransGen, Beijing, China. The extracted proteins were separated on 10 or 15 percent sodium dodecyl sulfate–polyacrylamide gels and then transferred onto polyvinylidene fluoride PVDF membranes. The membranes were incubated overnight at 4 degrees Celsius with the following primary antibodies: ACSL4 Abcam, GPx4 Abcam, FTH1 Abcam, COX2 Abcam, and beta-actin Sigma. Finally, the membranes were incubated with corresponding secondary antibodies for 90 min at room temperature, and densitometry measurements of the western blot bands were taken by using Image-Pro Plus 6.0 software Media Cybernetics, Rockville, MD, USA.
Real-Time PCR Total RNA was isolated from the kidney tissues with TRIzol Invitrogen, USA. First-strand cDNA was synthesized with a reverse transcription kit TaKaRa, Japan. The relative mRNA expression levels of the ferroptosis-related genes ACSL4 and IREB2 were assayed with a quantitative real-time PCR kit TaKaRa, Japan with glyceraldehyde 3-phosphate dehydrogenase GAPDH as the internal control.
The primers used in this study were as follows: ACSL4, TTGTATGCTGCCTGTCCACTTGTT forward primer and ATTCTCTTTGCCATAGCGTTTTTCT reverse primer; IREB2, ATTCTGCCTTACTCAATACGGGTCC forward primer and ATTGCTTTGTTTGGTTTCCAGTCC reverse primer; and GAPDH, CGTGTTCCTACCCCCAATGT forward primer and TGTCATCATACTTGGCAGGTTTCT reverse primer.
Statistical Analysis All values are expressed as the mean plus or minus standard deviation SD. Statistical analyses were performed by one-way ANOVA followed by the appropriate tests with the statistical analysis program GraphPad Prism 6.0 GraphPad Prism Software, CA, USA. A P value less than 0.05 was considered to indicate a statistically significant difference.
Results
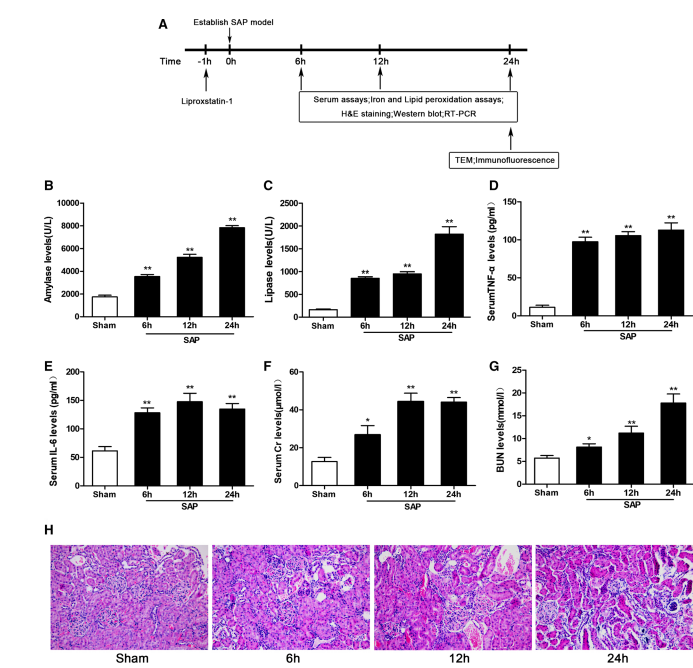
Successful Establishment of the SAP-Induced AKI Model After SAP induction, all the serum indexes changed significantly and continued to increase with time. As shown in Figure 1a–f, the levels of serum AMY, LPS, TNF-alpha, IL-6, Cr, and BUN were significantly higher in the SAP group at all time points than in the sham group. Compared with the sham group, tubular epithelial cell swelling, brush border loss, interstitial edema, tubular lumen obstruction, and cell necrosis were observed in the SAP group Figure 1h. These changes showed that our SAP-associated renal damage model was successfully established. Ferroptosis Is Induced in the Kidney After SAP To evaluate ferroptosis sensitivity in the context of SAP-induced kidney injury in rats, we assayed iron, MDA, and GSH levels and GPX4 activity in renal tissue. Iron, an essential factor for ferroptosis execution, accumulated significantly in the SAP group compared to the sham group Figure 2a. In addition, the content of MDA in renal tissue increased significantly, while the levels of GSH and GPX4 activity decreased Figure 2b–d.
Fig. 1 Serum biochemical indicators, inflammatory cytokines, and renal histological changes at different time points.(a) Experimental design: Lip-1 was administered 1 hour prior to the induction of severe acute pancreatitis (SAP). Animals were sacrificed for analysis at 6 h, 12 h, and 24 h post-SAP induction.(b–g) Serum levels of AMY (b), LPS (c), TNF-α (d), IL-6 (e), creatinine (Cr, f), and blood urea nitrogen (BUN, g) were significantly increased in the SAP group compared to the sham group.(h) Representative hematoxylin and eosin (H&E)–stained sections of kidney tissue (original magnification ×200) showing SAP-associated renal damage.Data are presented as mean ± SD (n = 8).* p < 0.05, **p < 0.01 vs. sham group.These findings confirm the successful establishment of a SAP-associated renal injury model.
Some core factors, such as ACSL4, GPX4, and FTH1, are accepted as pivotal ferroptosis-regulating proteins. Therefore, we determined the expression levels of these proteins. As shown in Figure 3a–d, the expression of the positive regulator ACSL4 was induced in injured renal tissue compared with normal tissue. In addition, the levels of two negative regulators, GPX4 and FTH1, were reduced. We also assessed the mRNA levels of the ferroptosis-related genes ACSL4 and IREB2 in the kidney. The mRNA levels of these genes were significantly higher in the SAP group than in the sham group Figure 3e, f. The results described above showed that the difference was the most prominent in the 24 h SAP subgroup, so we speculate that ferroptosis is likely more active at 24 h than at the other times investigated.
Immunofluorescence microscopy showed that ACSL4 red and GPX4 red were mainly expressed in renal tubular epithelia Figure 4a. The change is consistent with that of Western blotting. Next, TEM was used to investigate the morphological features of ferroptosis, and obvious mitochondrial shrinkage in the soma was observed in the SAP group Figure 4b. These findings indicate that SAP induces ferroptosis in the kidney, suggesting that ferroptosis may be involved in SAP-associated renal damage.
Lip-1 Inhibits Ferroptosis Effectively Lip-1, a potent and specific ferroptosis inhibitor, has previously been shown to attenuate renal I/R damage. As shown in Figure 5a–c, 24 h after induction of the SAP model, Lip-1 decreased lipid peroxidation, as indicated by the decrease in MDA level and the increases in GSH levels and GPX4 activity. Furthermore, Lip-1 treatment 1 h before SAP reduced ACSL4 expression and induced GPX4 and FTH1 expression Figure 3a–d, and the expression of ferroptosis-related genes was significantly lower in the SAP + Lip-1 group than in the SAP group Figure 3e, f. There was a marked change in ACSL4 red and GPX4 red in Lip-1-treated rat, as indicated by immunofluorescence Figure 4a. In addition, the morphological characteristics of mitochondria were also significantly ameliorated in the SAP + Lip-1 group Figure 4b. The changes in these indexes indicate that Lip-1 treatment effectively inhibits ferroptosis in renal injury with SAP.
Inhibition of Ferroptosis Attenuates SAP-Induced Kidney Injury To further verify the existence of ferroptosis and its role in SAP-induced kidney injury and to determine whether inhibition of ferroptosis could resolve kidney injury, we observed serum biochemical indicators and histopathological changes after 24 h of SAP. As shown in Figure 6a, b, serum AMY and LPS levels were significantly lower in the SAP + Lip-1 group than in the SAP group. Moreover, the levels of TNF-alpha, IL-6, Cr and BUN were significantly reduced in the SAP + Lip-1 group Figure 6c–f.
H&E staining was used to evaluate pancreatic and renal histological changes Figure 6g, i. No obvious pathological changes were found in the pancreas and kidney in the sham group. The histological scores of pancreas and kidney injury were markedly elevated in the SAP group compared with the sham group. However, the pancreatic histological scores in SAP rats were significantly ameliorated by pretreatment with Lip-1 Figure 6h. Furthermore, Figure 6i shows that SAP rats demonstrated glomerular and tubular damage as well as inflammatory cell infiltration. However, compared with SAP rats, rats pretreated with Lip-1 demonstrated fewer histological features and lower scores in the kidney Figure 6i, j.
Collectively, our findings demonstrate the involvement of ferroptosis in SAP-induced AKI and reveal that Lip-1 attenuates renal injury and restores renal function by inhibiting ferroptosis.
Discussion
In the present study, we found for the first time that ferroptosis, an iron-dependent form of programmed cell death, plays a critical role in renal damage associated with SAP. Moreover, we observed that SAP-associated oxidative stress, renal dysfunction and histological damage can be ameliorated by inhibition of ferroptosis.
AKI is one of the most common complications increasing the mortality rate of patients with SAP. Understanding the molecular mechanisms involved in renal tubular epithelial cell death is of great importance for the prevention of SAP-induced renal failure. To investigate these mechanisms, we established a model of SAP with AKI in rats. In the present study, we observed iron accumulation and increased lipid peroxidation in renal tissue of rats with SAP; these changes were accompanied by reductions in GPX4 activity and upregulation of ferroptosis-related proteins and genes, consistent with the occurrence of ferroptosis. Decreased expression of GPX4, which is critical for ferroptosis execution, was responsible for the induction of ferroptosis, because GPX4 inhibition invariably causes ferroptosis. As immunofluorescence microscopy showed that ACSL4 and GPX4 were mainly expressed in renal tubular epithelia, we speculate that ferroptosis mainly occurs in renal tubular epithelia. In addition, using TEM, we identified shrunken mitochondria in renal tubular epithelial cells 24 h after SAP. Furthermore, administration of Lip-1, which can specifically inhibit ferroptosis, not only decreased inflammatory factor levels and lipid peroxidation but also ameliorated renal tissue damage and improved renal function. In summary, these findings suggest that ferroptosis occurs in this rat model of SAP-associated renal damage and might thus be a therapeutic target for SAP-induced AKI.
At present, the pathogenesis of SAP-induced renal injury remains elusive. It is generally recognized that excessive release of cytokines and inflammatory mediators plays a pivotal role in SAP and SAP-associated renal injury. Previous studies have shown that SAP-related renal injury is caused mainly by oxidative stress-induced renal tubular epithelial cell barrier loss and dysfunction. In addition, other forms of cell death, including necrosis, autophagy, and apoptosis, were also observed in AKI, suggesting that renal damage may represent a combination of several different forms of cell death. However, the role of ferroptosis in SAP-induced AKI has not been reported. Our study indicates that ferroptosis plays an important role in AKI associated with SAP and suggests that it could be a lethal process induced by inflammatory mediators and oxidative stress. As ferroptosis is a dynamic process, we observed the expression of ferroptosis-related molecules at different time points and found that ferroptosis is likely most active 24 h after SAP. Undoubtedly, SAP-induced renal injury involves complex cell death mechanisms. However, the orchestrated connections among these cell death processes need to be studied in greater depth.
Ferroptosis is a newly discovered form of RCD that has been implicated in several pathophysiological contexts, such as IRI, tumor suppression, degenerative diseases, stroke, and antiviral immunity. Lip-1 potently and specifically inhibits ferroptosis by scavenging ROS in vivo. Recent studies have indicated that inhibition of ferroptosis alleviates rhabdomyolysis-associated renal damage in rats. In our current study, the levels of lipid peroxidation and renal injury were attenuated in SAP animals that had been treated with Lip-1.
In conclusion, our study provides significant evidence to confirm the occurrence of ferroptosis in an animal model of SAP-induced AKI and demonstrates that inhibition of ferroptosis can ameliorate renal dysfunction and histological damage. These findings reveal a new form of cell death that occurs in the context of SAP-induced AKI and provide a novel therapeutic target for renal injury.