
Selectivity Profile of Ro-3306: Evaluating Pharmacological Inhibitors of Cell-Cycle CDKs
ABSTRACT
Cyclin-dependent kinases (CDKs) are an important and emerging class of drug targets for which many small-molecule inhibitors, such as Ro-3306, have been developed. However, there is often insufficient data available on the selectivity of CDK inhibitors (CDKi) to attribute the effects on the presumed target CDK to these inhibitors. Here, we highlight discrepancies between the kinase selectivity of CDKi and the phenotype exhibited; we evaluated CDKi (claimed to target CDK1−4) for activity toward CDKs and for effects on the cell cycle. Our results suggest that most CDKi should be reclassified as pan-selective and should not be used as a tool. In addition, some compounds did not even inhibit CDKs as their primary cellular targets; for example, NU6140 showed potent inhibition of Aurora kinases. We also established an online database of commercially available CDKi for critical evaluation of their utility as molecular probes. Our results should help researchers select the most relevant chemical tools for their specific applications.
CDK1 Inhibitors: RO-3306 Is the Most Suitable CDK1 Chemical Probe to Date. The strong reported CDK1/B inhibition by the purine-based CGP74514A (published IC50 = 31 nM) led to a simple classification of this inhibitor as a CDK1-selective without any further investigation of other possible targets. The reported G2/M cell cycle arrest is partly in line with the expected target, but our own results (the G2/M-arrested population increased only by 19% compared to control after 24 h treatment with 5.9 μM compound) indicated that CDK1 is unlikely to be the sole target (Figure 2A). Indeed, our biochemical assays revealed that this inhibitor primarily targets CDK2 rather than CDK1 (Figure 3). These results are in agreement with broad kinase selectivity profiling using a radiometric filter-binding assay. CGP74514A was tested at three concentrations against >300 kinases, and the results obtained indicate that it exhibits a pan-selective pattern of inhibition. Interestingly, screening based on a thermal shift assay revealed that CGP74514A has a preference for group II of p21-activated kinases (PAKs), which are known nonspecific targets of purine-based CDK inhibitors. Despite the availability of sufficient information about CGP74514A targeting CDK2, it has been used several times as a CDK1-specific chemical probe. Some authors were aware that the identity of the cellular target of this inhibitor was unclear and therefore compared CGP74514A with Ro-3306, another well-established CDK1 inhibitor.
INTRODUCTION
A substantial fraction of proteins are modified by phosphorylation catalyzed by protein kinases,1 which represent the largest group of cellular targets for directed anticancer therapy. The human genome encodes 518 protein kinases, which regulate most cellular processes. However, the complexity of protein phosphorylation networks makes experimental studies aimed at dissecting the functions of individual kinases and identifying kinase substrates challenging. The use of various approaches such as RNA interference, chemical inhibitors, in vivo knockout mouse models, or CRISPR/Cas9 technology often leads to discrepancies or contradictions.
An important criterion for a good chemical probe is biological selectivity for the principal target. While the range of chemical probes targeting several protein kinases appears to be sufficient, with dozens of commercially available inhibitors for certain kinases, the selectivity information available is often incomplete or hard to find. Cyclin-dependent kinases (CDKs), together with other cellcycle protein kinases (Aurora, polo-like, and checkpoint kinases), have been well validated as targets for cancer treatment; three inhibitors have already been approved as drugs. The human CDK family comprises 20 members; some members have been described as essential regulators of the cell cycle (CDKs 1, 2, 4, 6), while others are involved in diverse cell-cycle-independent processes, such as regulation of transcription (CDKs 7, 8, 9, 11, 12), splicing (CDK12), DNA repair (CDKs 2, 9, 12), migration and angiogenesis (CDK5), or spermatogenesis (CDK16). In addition, most CDKs have numerous substrates and often participate in quite distinct processes, such as CDK7 (serving as both a CDK-activation kinase and an activator of RNA polymerase II), CDK5 (regulating neuronal function and cell migration), CDK9 and CDK12 (activation of RNA polymerase II, DNA damage response), and CDK6 (cell cycle, kinase-independent transcriptional regulation). Nevertheless, the precise biological function of some CDKs and cyclins has yet to be convincingly established.
Intensive research in the development of CDK inhibitors (CDKi) has led to the identification of many compounds that differ in selectivity toward members of the CDK family and in potency and cellular effects. Currently, over 100 different CDKi are commercially available for experimental work. These CDKi are often classified according to their selectivity as described in the original literature, but in some cases the selectivity has not been well studied. The classification of compounds discovered 10−20 years ago may be incorrect due to the limited knowledge of other CDKs at that time; many such “selective” CDK inhibitors were assayed against at most 3 kinases out of 20 (Figure 1). Some CDKi were classified as pan-selective (i.e., inhibiting most CDKs), whereas the rest were classified as selective inhibitors for a single CDK or two specific CDKs (usually referred to as dual inhibitors). The latter classification, in particular, merits closer inspection because in many cases there is little obvious mechanistic basis that could explain dual selectivity. In fact, in-depth characterizations of the selectivity of many such compounds have often been published later. However, these characterizations have often been part of large studies addressing several kinase inhibitors and are not easy to find in the literature. Importantly, commercial catalogues frequently do not reflect these updates, and if a scientist relies solely on information provided in the product datasheet, an unsuitable probe could easily be selected and purchased.
Figure 1. Histogram showing the percentage of tested “selective” CDK inhibitors against specified number of CDK/cyclin complexes. Formerly, 52% of “selective” CDK inhibitors (62 inhibitors included) were assayed against at most 3 CDK complexes out of 20 (only IC50, Ki and Kd data included) (white bars) while after complementation with our data (gray bars), 61% of CDK inhibitors were characterized by at least 7 CDK/cyclin complexes inhibited. Information about classification, alternative names, CAS numbers, kinase inhibition data, and references of inhibitors are available in the Supporting Information, File S1.
Our aim was to provide more information about inhibitors targeting cell-cycle CDKs that we considered to be insufficiently validated and to increase the available knowledge about these compounds as chemical tools; to do so we evaluated these inhibitors in terms of activity against a panel of CDKs 1, 2, 4, 5, 7, and 9 (Figure 1) and effects on the cell cycle of HCT-116, a cell line commonly used for phenotypic screen of cell-cycle modulators. We did not evaluate inhibitors that display a pan-selective pattern of inhibition or whose inhibitory preferences for CDKs have been described in detail elsewhere (such as the clinically developed dinaciclib, SNS032, roscovitine, flavopiridol, and AT7519, which are also less selective). Our study further highlights discrepancies between kinase selectivity and the phenotype exhibited. These results should help researchers select the most relevant chemical tools for their specific applications.
RESULTS
Figure 2. (A) Effect of inhibitors designated CDK1-selective on the cell cycle of HCT-116 cells treated for 24 h: flow cytometric analysis (10 000 counts) of DNA stained with propidium iodide. (B) Immunoblot confirming thermal stabilization of CDK1 protein levels by Ro-3306 in HCT-116 cells after 4 h of treatment.
Figure 3. (A) Structures of inhibitors designated as CDK1 selective. (B) Kinase inhibition data expressed as IC50 values complemented by graphic illustration of the selectivity for certain CDK. Red and green bars indicate CDK activity on a log10 scale (midpoint corresponds to 1 μM, and maximum for red and green is 1 nM and 100 μM, respectively). Cellular phenotype is determined in HCT-116 cells treated for 24 h.
“CDK1 inhibitor” (compound 8a; published IC50 = 5.8 μM), was developed from a library of 2-indolinone derivatives based on the predicted binding to the homology model of CDK1; however, further kinase inhibition or biological data for this compound have not been published. Our measurement confirmed its micromolar potency toward CDK1 but not the selectivity (Figure 3). Published data from a broad-spectrum kinase profiling of “CDK1 inhibitor” also revealed nonselective inhibition of several kinases; importantly, CDK1 is not present among the most sensitive kinases (CDK1 ranks 103rd out of 292). Nevertheless, this compound arrested HCT-116 cells in the G2/M phases (Figure 2A), but due to poor potency and due to the nonselective nature of “CDK1 inhibitor”, the underlying mechanism for this arrest might not be the inhibition of CDK1. Another possible target is MLCK (11% residual activity with 10 μM compound), the inhibition of which can cause similar mitotic defects and cytokinesis failure
The 1-aza-9-oxafluorenes benfluorene and elbfluorene have also been designated as CDK1-selective inhibitors. The more effective elbfluorene inhibits CDK1 (published IC50 = 4.2 μM), while inhibition of CDK2 and CDK4 was not observed at up to 100 μM. However, our measurement did not confirm inhibition of CDK1 or of any other tested CDKs (Figure 3). In addition, we did not observe any effect on the cell cycle profile of the inhibitor-treated HCT-116 cells (Figure 2A). The thiazolinone derivative RO330632 is probably the most frequently used chemical tool in studies investigating the role of CDK1. Surprisingly, the selectivity profile of Ro-3306 has only been partially revealed; a recent article described Ro-3306 potency only toward CDK1 and CDK2,35 and partial kinase profiling data showed that CDK2/A and CDK9/T were only weakly inhibited. Nevertheless, published data demonstrate that Ro-3306 can also effectively inhibit other kinases from the CMGC group of kinome, especially DYRKs. We complemented this kinase profile and confirmed the high selectivity of Ro-3306 for CDK1 (Figure 3). Consistent with potent CDK1 inhibition, Ro-3306 treatment strongly arrested the cell cycle of the HCT116 cell line in the G2/M phase (Figure 2A). This result has been described in many studies on different cell lines as usually leading to a substantial G2/M block or to cyclin B accumulation, which clearly corresponds to the inhibition of CDK1 at the cellular level.
To further confirm the direct interaction of studied inhibitors with CDK1, we performed a cellular thermal shift assay (CeTSA), which is based on the ligand-induced thermal stabilization of target proteins of HCT-116 cells treated with different concentrations of compounds that affect the cell cycle of these cells. Our data clearly showed that, in contrast to CGP74514A and “CDK1 inhibitor”, only Ro-3306 can stabilize CDK1 expression levels in inhibitor-treated cells (Figure 2B). The above-mentioned results strongly highlight Ro-3306 as the most suitable tool for pharmacological inhibition of CDK1, while CGP74514A should be classified as a pan-selective inhibitor. “CDK1 inhibitor”, benfluorene, and elbfluorene were identified as being inactive toward CDKs.
CDK2 Inhibitors: NU6140 Strongly Inhibits Aurora Kinases in Vitro. Inhibitors that have been claimed to exhibit selectivity for CDK2 are the most numerous; however, most of them are designated as dual inhibitors due to the strong activities of these compounds toward the structurally related CDK1 and in some cases toward CDK5 and CDK9. Purine derivatives CVT313 (marketed as “CDK2 inhibitor III”) and NU6140 (marketed as “CDK2 inhibitor IV”) inhibit CDK2 in a nanomolar range and exhibit selectivity over CDK1 and transcriptional CDKs (Figure 4). Nevertheless, we found that CVT313 also interacts with recombinant CDK5 (Figure 4), which is in line with published kinase profiling. In addition, cellular inhibition of CDK5 with CVT313 was confirmed by monitoring of phosphorylated FAK at S732 that Information, File S2). Both the purines arrested HCT116 cells in the G2/M phase, but NU6140 treatment resulted in a significant tetraploid population (Figure 5A). This observation led us to hypothesize that the main target of NU6140 might not be CDK2 (determined IC50 = 0.4 μM) but another mitotic kinase, such as PLK or AURs. The screening data for NU6140 against a panel of 300 kinases suggest a possible explanation. While CDK2 was only weakly inhibited by this compound (CDK2 ranked 54th out of 300; 500 nM NU6140 caused 44% inhibition), all three Aurora kinases were substantially more sensitive (2nd, 16th, and 23rd ranks). We therefore determined the IC50 for AURs and found that the values were approximately 6−10 times lower than that for CDK2 (Figure 5B). To further confirm cellular inhibition of AURB, we performed immunoblotting analysis to examine phosphorylated histone H3, a direct substrate of AURB. NU6140 indeed decreased phosphorylation of H3-S10 in HCT-116 cells in a dose-dependent manner (Figure 5C). In addition, the structurally related 2,6-disubstituted purine reversine was described recently as an inhibitor of AURs. Our findings thus provide novel insight into the possible mechanism of action of NU6140.
Figure 4. (A) Structures of inhibitors designated as CDK2 selective. (B) Kinase inhibition data expressed as IC50 values complemented by graphic illustration of the selectivity for certain CDK. Red and green bars indicate CDK activity on a log10 scale (midpoint corresponds to 1 μM; maximum for red and green is 1 nM and 100 μM, respectively). Cellular phenotype is determined in HCT-116 cells treated for 24 h.
Figure 5. (A) Effect of different concentrations of NU6140 on the cell cycle of HCT-116 cells treated for 24 h: flow cytometric analysis (10 000 counts) of DNA stained with propidium iodide. (B) Inhibition of Aurora kinases by different CDK inhibitors. JNJ7706621 (dual CDK/AUR inhibitor) and tozasertib (AURs inhibitor) were used as positive controls. (C) Immunoblotting analysis of Aurora kinases and phospho-histone H3 in HCT-116 cells treated with NU6140 for 24 h. β-Actin was used as a loading control.
Figure 6. (A) Structures of inhibitors designated as CDK4 selective. (B) Kinase inhibition data expressed as IC50 values complemented by graphic illustration of the selectivity for certain CDK. Red and green bars indicate CDK activity on a log10 scale (midpoint corresponds to 1 μM; maximum for red and green is 1 nM and 100 μM, respectively). Cellular phenotype is determined in HCT-116 cells treated for 24 h.
Indolinone derivatives GW8510 and “CDK2 inhibitor II” exhibit nanomolar potency for CDK2 (published IC50 = 10− 60 nM). We found here that “CDK2 inhibitor II” has higher selectivity for CDK2 than for other members of the CDK family, whereas GW8510 showed strong activity against CDK5 and CDK1 (IC50 ≈ 7 and 49 nM, respectively) (Figure 4) that was confirmed by dephosphorylation of FAK at S732 (CDK5 substrate) and durable G2/M arrest in treated CDK2 knockout HCT-116 cells. Both inhibitors increased the G2/M-arrested population of HCT-116 cells. Unfortunately, usage of the more selective “CDK2 inhibitor II” as a chemical probe is limited due to its poor solubility. Despite this limitation, in recent articles, “CDK2 inhibitor II” was used as a tool to highlight the role of CDK2 in the promotion of tumor proliferation and induction of radio resistance in glioblastomas and in the phosphorylation of ligand-dependent progesterone receptor at S400,50 histone methyltransferase Suv39H1 at S391,51 and enhancer of zeste 2 (EZH2) at T416.52 However, results of these studies should be interpreted with caution. First, the concentration range of inhibitor that was used to determine these effects was broad (70 nM to 4 μM). Second, profiling of a broader panel of kinases has not been reported. Lastly, many related indolinone derivatives are less selective.
The indolinone SU9516 is one a few inhibitors that have been well-profiled, including against different CDKs. However, the preference of this inhibitor for CDK2 is not robust; our data revealed similar activity against CDK5/p25 (Figure 4) that was confirmed also in cells by dephosphorylation of pFAK(S732). This finding is in agreement with reported profiling, where CDK5 was seen to be the most sensitive out of 300 tested kinases (Supporting Information, File S2).12 Cell cycle analysis of inhibitor-treated HCT-116 cells indicated G2/M arrest (Supporting Information, Figure S3), which is consistent with data on other cell lines, namely, T24, SPC-A1, or RKO. Moreover, significant G2/M block in SU9516-treated HCT116 cell lacking CDK2 showed that SU9516 also targets CDK1 in cells.
Figure 7. Cellular effects of commercially available CDK inhibitors marketed as “CDK4 selective” or “dual” do not always correspond to expected phenotype. Shown are cell cycle analyses of HCT-116 cells treated for 24 h with CDK inhibitors from the same group (panel A, marketed as CDK4 selective; panel B, designated dual inhibitors). Shown is flow cytometric analysis (10 000 counts) of DNA stained with propidium iodide.
Milciclib (PHA848125) was reported to uniquely inhibit CDK2/A with 10-fold higher potency than for CDK2/E (published IC50 = 45 nM and 363 nM, respectively); however, due to nanomolar inhibition of tropomyosin receptor kinases (TRKs), milciclib has also been designated a dual CDK-TRK inhibitor. Our measurements confirmed the published data but revealed that milciclib can also moderately inhibit CDK4 (Figure 4). This finding is probably associated with the structural similarity of this compound to the CDK4 selective palbociclib (PD0332991) and ribociclib (LEE011). Cellular inhibition of CDK4 is evident from the analysis of the cell cycle, especially at low inhibitor concentrations, which arrest HCT-116 cells in the G1 phase. Conversely, higher doses lead to G2/M arrest, probably due to effective inhibition of CDK2. Similar effects were also reported with ovarian cell lines.
Our evaluation showed that no commercially available inhibitor designated as CDK2 selective should be used as a CDK2 probe in cellular experiments. The compounds have either low solubility (“CDK2 inhibitor II”), low CDK selectivity (GW8510, SU9516, CVT313), or heterogeneous cellular effect (milciclib and NU6140). Clinical CDK4/6 Inhibitors Are Selective Chemical Tools. CDK4 and CDK6 exhibit certain structural differences in comparison with other CDKs, and inhibitors of these kinases are usually much more selective than other CDKi.59 This selectivity probably contributed to the successful approval of palbociclib (PD0332991), ribociclib (LEE011), and abemaciclib (LY2835219) for the treatment of advanced breast cancer. Although these drugs share some pharmacophores (Figure 6), recent data pointed at some distinctions that confirmed their differences related to their off-targets. Palbociclib, but not ribociclib, has been shown to specifically interact with several lipid kinases PIP4K2A/B/C and to increase the number of autophagic vesicles via inhibition of AKT signaling in lung cancer. Autophagy induced by palbociclib was observed also in hepatocellular carcinoma via the PP5/AMPK axis, while ribociclib and abemaciclib had minimal effects in this model. Further study revealed that in contrast to palbociclib and ribociclib, abemaciclib directly inhibits GSK3α/β and CAMKIIγ/δ kinase activity and potently activates β-catenindependent WNT signaling. Non-kinase binding studies and kinome interaction analyses revealed that abemaciclib inhibits also CDK9,13 but cellular studies did not find any changes in phosphorylation of CTD RNA polymerase II (a CDK9dependent process) in different cell lines.69,70 And importantly, unlike palbociclib and ribociclib, abemaciclib exhibits cellular toxicity also in Rb-deficient cell lines in vitro, highlighting the possibility of having different targets.
We also performed the cell cycle analysis of inhibitor-treated HCT-116 to confirm the selectivity of these CDK4/6 inhibitors and expected phenotype (G1 block without significant apoptosis in Rb-positive cell lines). As we supposed, palbociclib and ribociclib arrested the cell cycle in G1 phase in nanomolar concentrations. Micromolar concentrations of palbociclib and abemaciclib, but not ribociclib increased G2/M population, which is in agreement with previous reports. Nevertheless, micromolar concentrations of these drugs do not fall within therapeutically relevant doses, and therefore only concentration of <1 μM should be used when targeting CDK4/6 probe. Mostly palbociclib and ribociclib have frequently been used as chemical probes in numerous biological studies; however, other “CDK4/6-selective” inhibitors are also commercially available. NSC625987 (sold as “CDK4 inhibitor II”)76 is a thioacridone derivative of 3-amino-9-thio(10H)-acridone (3ATA), one of the first inhibitor published as having a preference for CDK4 over CDK1/2. Nevertheless, our measurements did not reveal any affinity to the assayed CDKs at concentrations up to 20 μM (Figure 6). Our findings are consistent with published kinase profiling data (Supporting Information, File S2), in which none of the 234 tested kinases were inhibited with more than 50% efficacy by 10 μM NSC625987. In addition, our cell cycle analysis in inhibitor treated cells reveal only moderate G1 phase arrest accompanied by massive apoptosis, further disqualifying NSC625987 from being a suitable chemical probe for CDK4.
Figure 8. (A) Structures of inhibitors showing preference for two CDKs (designated as dual). (B) Kinase inhibition data expressed as IC50 values complemented by graphic illustration of the selectivity for certain CDK. Red and green bars indicate CDK activity on a log10 scale (midpoint corresponds to 1 μM; maximum for red and green is 1 nM and 100 μM, respectively). Cellular phenotype is determined in HCT-116 cells treated for 24 h.
Ryuvidine (“CDK4 inhibitor III”), a derivative of benzothiazole, was reported to weakly but selectively inhibit CDK4 (published IC50 = 6 μM); however, there are no published results that show cellular inhibition of CDK4 by this compound. Our measurement confirmed micromolar activity toward CDK4, but due to the limited solubility of this inhibitor, we could not record IC50 value with other CDKs (IC50 > 20 μM) (Figure 6). Published kinase profiling of ryuvidine revealed that none of the 300 kinases tested were inhibited with more than 40% efficacy. Our cell cycle analysis did not show G1 arrest in inhibitor-treated HCT-116 cells (Figure 7); however, a rather unexpected accumulation of cells in the S phase was observed (increased by 15% compared to the control), which is not typical of CDKi and therefore suggests inhibition of other targets. Consistent with this finding, recent work has shown that ryuvidine strongly inhibits the lysine methyltransferase protein SETD8 (IC50 = 0.5 μM), inhibition of which results in cell cycle defects in the S and G2/M phases.
The compound CINK4 (available as “CDK4/6 inhibitor IV”) was reported to inhibit CDK4 in micromolar ranges; however, our data also revealed measurable inhibition of CDK1 and CDK2 (Figure 6), discouraging its use as a specific probe. Broader selectivity toward CDKs was further confirmed by flow cytometric analysis of inhibitor-treated HCT-116 cells; we documented significant G1 arrest upon treatment with lower doses of this compound but clear G2/M arrest with higher doses (>10 μM) (Figure 7). Indolocarbazoles arcyriaflavin A81 and its brominated derivative sold as “CDK4 inhibitor” are structurally related to the multikinase inhibitor staurosporine. Our data confirmed the preference of arcyriaflavin A and “CDK4 inhibitor” for CDK4 (Figure 6), but both compounds also inhibited CDK2 and CDK5 at nanomolar levels (Figure 6). Published results for “CDK4 inhibitor” from broad-spectrum kinase profiling showed targeting of CDK4 and CDK6 (9th and 5th rank, respectively, among the 300 kinases tested) but also confirmed nonspecificity throughout the kinome.12 Cell cycle analyses confirmed the pan-selectivity of both compounds; while low doses of inhibitor lead to G1 arrest of HCT-116 cells, higher doses cause accumulation of cells in G2/M phases. This finding has also been documented for other indolocarbazole derivatives. On the basis of our investigation, we cannot recommend the use of these two inhibitors as specific chemical tools.
Isoquinolinone-based “CDK4 inhibitor V” is described as a selective nanomolar inhibitor of CDK4; however, this description is not consistent with our measurements. While we indeed confirmed nanomolar inhibition of CDK4 (IC50 = 38 nM), we found that this compound also inhibits CDK1 and CDK2 at a submicromolar range (Figure 6). Analysis of the effects of this compound on the cell cycle further confirmed our findings; treatment of HCT-116 cells with this inhibitor resulted in G2/M arrest of cell cycle (Figure 7). ON123300, which is structurally related to palbociclib, is known for its preference for not only CDK4 (published IC50 = 4 nM) but also AMPK-related protein kinase 5 (ARK5) (published IC50 = 4 nM). Our data revealed that ON123300 is also able to strongly inhibit CDK2 (Figure 6). An additional biochemical screening revealed that ON123300 is a nanomolar inhibitor of other kinases such as FLT3, FYN, Abl, and PDGFRβ. When used at nanomolar doses, the effect of this compound on HCT-116 cells is associated with the inhibition of CDK4; however, using higher doses (>1 μM) leads to the accumulation of cells in the G2/M phases, which was also independently reported in lymphoma cells. Taken together, our analysis revealed that only few compounds (palbociclib, ribociclib, and abemaciclib) cause typical CDK4-specific cellullar phenotype (G1 block without induction of apoptosis) in treated cells in a dose−response manner, whereas others display lower CDK4/6 selectivity (ON123300, “CDK4 inhibitor”, CINK4, arcyriaflavin A, and “CDK4 inhibitor V”) or probably another mechanism of action (NSC625987 and ryuvidine).
Dual Inhibitors: More than Just Two Targets. We were further interested in CDKi that were classified as dual inhibitors, suggesting lower selectivity; such compounds should be avoided by researchers who need a selective chemical tool. Our results actually provide evidence that none of the studied compounds selectively inhibit only two CDKs in biochemical assays (Figure 8). Moreover, only three of them, namely, BMS265246,86 “CDK2/9 inhibitor”, and BML259, exhibit the highest potency for the two CDKs that are designated as the major targets of these inhibitors. We found that some other CDKs are inhibited nearly to the same extent (e.g., the “CDK2/9 inhibitor” also inhibits CDK5). All these compounds, together with “CDK1/2 inhibitor III” and CGP60474, should be reclassified as pan-selective.
Several other inhibitors were reported to effectively inhibit not only CDKs but also other kinases, which therefore raises concerns regarding their use as selective CDK probes. For example, the indirubin derivative BIO (6-BIO) inhibits glycogen synthase kinase-3β, while the purine (R)-DRF053 exhibits potent activity toward casein kinase CK1δ.93 “CDK1/ 5 inhibitor” and PNU112455A95 exhibit limited activity against CDKs.
To further verify the selectivity of investigated inhibitors, we performed immunoblot analysis of different CDK substrates (namely, pFAK-S732 and pRNA polymerase II as substrates of CDK5 and CDK7/9, respectively) in treated HCT-116 cells. Our results clearly showed that “CDK1/2 inhibitor III”, BMS265246, and CGP60474 (all designated as CDK1/2 inhibitors) also inhibit CDK5 in cells and confirmed results from biochemical assays. Oppositely, relatively high affinity of “CDK1/2 inhibitor III” and CGP60474 for transcriptional CDKs from kinase assays did not express significantly in the dephosphorylation of RNA polymerase II.
Further, we confirmed the preference of these inhibitors for CDK1 and CDK2 in cells; our data from flow cytometry on treated HCT-116CDK2−/− cells corresponded with expected phenotype that co-depletion of CDK1/2 together causes the greatest increases in G2/M cell cycle. In the same experiment we also revealed that CDK inhibitors declared to not inhibit other CDKs (“CDK2/9 inhibitor” and BML259) potently inhibit CDK1 and CDK5 in cells. NU6102 is a purine-based compound bearing an O6cyclohexylmethyl moiety and was developed by the optimization of NU6027 and NU2058 (also commercially available). NU6102 shows a binding mode different from other CDK inhibitors with a purine scaffold (such as roscovitine, CR8, or H717); one can therefore expect this compound to also exhibit substantially different potency and selectivity. NU6102 was formerly described as a nanomolar inhibitor of CDK1/2 (published IC50 = 9 and 6 nM), but additional studies revealed that NU6102 exhibits high selectivity for CDK2 over other CDK-family members, including CDK1.34,35,37,100 In addition, the selectivity of NU6102 in cells was confirmed by the differences observed in the cytotoxicity of NU6102 toward cells lacking CDK2 compared to wild-type cells, and further study showed that NU6102’s specifity over CDK1 in the Xenopus homologs of these kinases.
Our data confirmed the reported preference for CDK2 and the weak activity against transcriptional CDKs (Figure 8). Treatment of HCT-116 cells resulted in rapid G2/M arrest, as reported previously in other cell lines. Nevertheless, flow cytometric analysis of NU6102-treated HCT-116 cells lacking CDK2 revealed significant G2/M block suggesting CDK1 inhibition. Interestingly, HCT-116 cells with acquired resistance to NU6102 exhibit upregulated CDK6 activity, suggesting functional compensation between these two slightly divergent kinases. NU6102 has been further optimized to develop inhibitor, which is 2000-fold less active toward CDK1 (published IC50 = 86 μM) while retaining high potency against CDK2 (published IC50 = 0.044 μM). Unfortunately, this compound, which is the most CDK2-selective inhibitor to date, is not commercially available yet, and we could not include it in our study. Another structurally related and commercially available compound, NU6300, has been described as the first covalent CDK2 inhibitor.
To sum up, results from cellular experiments and biochemical profiling clearly discriminate the use of most dual CDKi as selective probes. Some of these CDKi indeed inhibit CDKs reported as main targets in nanomolar doses; nevertheless other CDK complexes are often inhibited to the same extent. Further, some inhibitors have preference for another cellular target (BIO and (R)-DRF053) or display poor potency (“CDK1/5 inhibitor”, PNU112455A) and one should avoid their application because it would be unreliable to confidently address CDK inhibition in cells. Cyclin-Dependent Kinase Inhibitor Database. In order to facilitate access to critical information about the utility of commercial CDK inhibitors as molecular probes, we created the cyclin-dependent kinase inhibitor database (CDKiDB) (http://rustreg.upol.cz/CDKiDB). At present, it comprises compounds identified by a survey of vendors’ catalogues. The following information about the individual inhibitors was compiled from original articles or reviews: chemical structure, CAS numbers and synonyms, SMILES, activities against the individual CDKs, total number of studied kinases, and references. The inhibitors are classified according to their CDK preference(s) given in literature and/or by vendors. We provide a critical comment on this classification for 31 inhibitors evaluated in this study. We encourage readers to join our effort to compile such information from both literature and unpublished data.
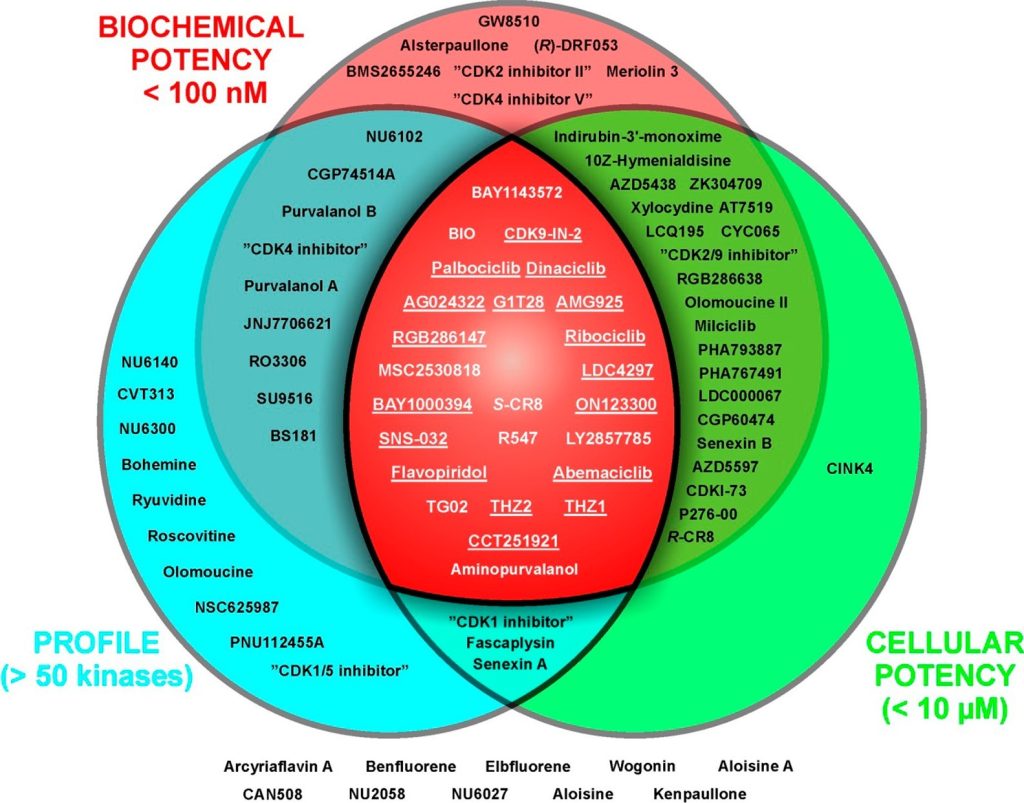
Figure 9. Distribution of commercially available CDK inhibitors according to their fitness factors,4 including biochemical potency (availability of IC50, Ki, or Kd values for at least one CDK), cellular potency related to direct inhibition of CDK/s in cells (e.g., dephosphorylation of retinoblastoma protein or RNA polymerase II (for CDK7 and CDK9 inhibitors), cell cycle arrest, BrdU incorporation or 7dF3 cell based assay (for CDK8/19 inhibitors), and selectivity profiling across the kinome. Underlined inhibitors also meet stricter criteria of cellular potency (<500 nM) and profiling on the panel of at least 125 kinases.106 Inhibitors K03861, CDK-IN-2, CDK9-IN-6, butyrolactone I, PHA690509, 3-ATA, “CDK1/2 inhibitor III”, BML259, WHIP180, CDK9-IN-2, indirubin, indirubin-5-sulfonic acid, 6-iodoindirubin-3′-monoxime, alosine B, and hymenidin are not included due to lack of information in one of the categories. Additional information about compounds, their alternative names, CAS numbers, kinase inhibition data, and references are available in the Supporting Information, File S1.
DISCUSSION AND CONCLUSION
Currently, about 100 CDK inhibitors of rather diverse chemical structures (for a dendrogram see Supporting Information, Figure S8) are commercially available, apparently providing a plethora of tools for chemical biology; however, the quality of these compounds, with respect to biochemical potency, selectivity, and expected cellular effects, can differ substantially. A useful chemical tool should meet several criteria, including a sufficient selectivity profile (>50-fold in biochemical assays of at least 125 kinases) and biochemical (<100 nM) and cellular (<500 nM) potency. These features have been defined as important “fitness factors” of chemical tools as defined recently. Unfortunately, many CDK inhibitors do not meet at least one of these factors (Figure 9). We have measured the inhibitory profile of 41 compounds (31 studied + 10 control CDK inhibitors) to gain information about their potency and selectivity of these compounds toward CDKs 1, 2, 4, 5, 7, and 9, and these results were further compared with data available in the literature. Furthermore, we analyzed the effects of inhibitors on the cell cycle in the HCT-116 cell line and attempted to link these effects to the biochemical profile.
We found that out of the 31 inhibitors studied, only palbociclib, ribociclib, and abemaciclib meet the above fitness factors and can be used as CDK4-selective probes; application of the other studied CDK4 inhibitors could be problematic. These results clearly reflect the clustering of CDK inhibitors according to CATDS (concentration- and target-dependent) score reported recently by Klaeger et al. Other candidates from different classes, namely, NU6102 and “CDK2 inhibitor II”, also showed good performance across two fitness factors, but the cellular potency was poor or limited. Nevertheless, only these two inhibitors exhibited reasonable selectivity for CDK2 and can be used as CDK2 probes. Alternatively, the irreversible CDK2 inhibitor NU6300 is available for studies. Although biochemical assays have revealed additional interactions with other kinases, there is little evidence of significant off-target activity of NU6300 in cells.104 Our comparison of the selectivity profiles of the tested CDK1 inhibitors clearly shows that Ro-3306 is the most suitable chemical tool; no other compound exhibited such high selectivity for CDK1 (∼4-fold). Furthermore, our results provide evidence that no dual inhibitor selectively inhibits only two of the assayed CDK complexes; they usually exhibit broader spectra of inhibition. We also showed that CVT313 and GW8510, both designated as CDK2 inhibitors, also exhibit high potency toward CDK5. The use of other compounds, such as “CDK1/5 inhibitor”, CINK4, benfluorene, elbfluorene, PNU112455A, and NSC625987, to study processes linked with CDKs should be strictly avoided due to the poor inhibition of CDKs by these compounds. Our CDK profiling has identified inhibitors that should be reclassified as pan-selective (e.g., CGP74514A, SU9516, and “CDK4 inhibitor V”). Moreover, some compounds did not inhibit CDKs as a main cellular target, despite original reports (e.g., “CDK1 inhibitor” and NU6140). While the major target of “CDK1 inhibitor” remains to be identified, our results provide evidence that NU6140 is a potent inhibitor of Aurora kinases (Figure 5).
Most CDKi also inhibit other protein kinases; in many cases, the compounds exhibit even higher affinity for these nonselective targets. Typical examples are BIO, ON123300, and milciclib, which are also potent inhibitors of GSK3, ARK5, and TRK, respectively. Kinome-wide selectivity data are unavailable for most compounds, and the lack of these data could be a factor that limits the use of these compounds in chemical biology. While some CDKi have indeed been profiled, the results are often buried in supplementary files of articles that do not focus directly on CDKs and are therefore hard to find. The available selectivity profiles could reveal potential nonspecific targets and thus improve the interpretation of obtained results; however, a lack of general awareness of the existence of these selectivity profiles seems to be a limiting factor. Additionally, these inhibitors are often assayed in single, sometimes suboptimal dose, which does not provide enough information and could limit the use of these compounds. For example, profiling of “CDK1/2 inhibitor III” was conducted at a 500 nM concentration, at which concentration more than half of the 300 kinases tested were inhibited with efficacy of >50%. With respect to the strong cellular potency, a lower concentration would be more suitable and would filter out weakly targeted kinases.
A literature survey revealed that at least 47 CDKi have been profiled against more than 50 kinases. These data revealed information about additional kinases that might also be inhibited in cells, which could therefore interfere with the interpretation of the results of cellular experiments. Typical examples include p21-activated kinases (PAKs) or casein kinases (CK1), which are inhibited by purine-based CDKi such as CGP74514A and (R)-DRF053. Notably, roscovitine and CR8 were initially believed to be pan-CDK inhibitors; however, subsequent studies have demonstrated that both these compounds also effectively inhibit CK1 and have been referred to as dual CDK/CK1 inhibitors.
In addition to nonspecific kinase targets, unrelated targets can also complicate the delineation of observed cellular effects. Non-protein kinase targets can be isolated, for example, using affinity chromatography on immobilized inhibitors; this approach led to the identification of pyridoxal kinase as a nonspecific target of roscovitine. Dinaciclib was found to interact with the bromodomain testis-specific protein BRDT, a member of the BET family of bromodomains, via a hinge-binding scaffold; however, this binding occurred at clinically irrelevant doses. Bromodomain-containing proteins might therefore be general targets of kinase inhibitors, as also shown recently for some clinically tested drugs, such as fedratinib and volasertib. Ryuvidine (“CDK4 inhibitor III”), one of the compounds studied in this paper, was shown in 2014 to inhibit the lysine methyltransferase SETD8 at clinically relevant doses. These examples highlight the possibility of obtaining misleading conclusions with nonselective inhibitors, as previously discussed. For example, SB203508 was originally developed as a p38α kinase inhibitor but was found to interact also with CK1.
Since most CDKs in our biochemical panel regulate the cell cycle, their selective inhibition should influence the distribution of cell populations in different phases. We therefore analyzed cell cycle effects in inhibitor-treated HCT-116 cells. A sharp G2/M arrest can indicate interactions with cell cycleregulated kinases;20 however, such a finding can sometimes be wrongly interpreted as a result of CDK1 inhibition. Only four of the studied compounds, including Ro-3306 and “CDK1/2 inhibitor III”, which are potent CDK1 inhibitors, were able to strongly (>80%) induce G2/M arrest (Figures 2, 5 and 7). In contrast, G2/M arrest caused by CDK1 inhibitors is probably not linked to CDK1 as the cellular effects were observed at doses much lower than the biochemical IC50 value of this compound. Surprisingly, the cellular effect of NU6140 seems to be related to the inhibition of Aurora kinases but not of CDK1 (Figure 5).
A majority of the tested inhibitors caused slight, but dosedependent, accumulation of cells in the G2/M phases, suggesting more complex cellular effects. Surprisingly, this observation was made not only for CDK2 or dual inhibitors but also for “CDK4 inhibitor V” and CGP74514A (a CDK1 inhibitor). Interestingly, some compounds induce dosedependent accumulation of cells in the G1 phase (up to a certain concentration), which is converted to a G2/M arrest upon treatment with higher doses. We assume that this effect is caused by strong cellular inhibition of CDK4 with low doses of compound, while higher doses also effectively inhibit CDK1 or CDK2. This phenomenon is observed for those CDKi that exhibit nanomolar affinity for CDK4 but do not have a high selectivity index, namely, ON123300, “CDK4 inhibitor”, CINK4, and milciclib.
It is evident that information about selectivity is crucial when choosing high-quality chemical tools, and it is important to avoid using nonoptimized and poorly profiled probes. Biochemical profiling assays with purified kinases should be complemented with in vitro phenotypic assays. Further profiling by different techniques, such as surface plasmon resonance, isothermal calorimetry, thermal denaturation assays, cellular thermal shift assays, microscale thermophoresis, mobility shift assays, and affinity chromatography coupled with proteomics, will contribute to the correct validation of these inhibitors as chemical tools and to the identification of possible nonspecific targets among unrelated proteins.
Last, but not least, instability of some (especially less explored) compounds may be another critical issue, which should be also considered. Some compounds could be chemically unstable in the assay media; possible modifications include redox reactions, hydrolysis, hydration, and isomerization. We could highlight for example benfluorene (an ethyl ester) and “CDK inhibitor II“, (a hydrazone), both susceptible to hydrolysis, or ryuvidine (1,4-quinone derivative), which may undergo redox reactions and serve as a dienophile for various Diels−Alder reactions. Consideration of chemical stability is therefore recommended to select reliable tool compounds and to produce high-quality data.
In conclusion, we created CDKiDB, an online resource for critical evaluation of commercial CDK inhibitors. It contains our commentary on the utility of the inhibitors based on the results presented in this paper. We plan to update and extend the evaluation by the results of follow-up studies. We would also like to encourage other researchers to contribute their data to the database.
EXPERIMENTAL SECTION
1. Cell Lines. The HCT-116 cell line (colorectal carcinoma) was obtained from the European Collection of Authenticated Cell Cultures. Briefly, cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/mL). MDA-MB-468 and HCT-116 (CDK2−/−) cells were kindly provided by Dr. Jan Bouchal from Department of Clinical and Molecular Pathology, Palacký University, in Olomouc, and Dr. Daniel Fisher from IGMM, CNRS, University of Montpellier, respectively. These cell lines were cultured in DMEM−high glucose medium supplemented with 10% fetal bovine serum and antibiotics. Cells were maintained in a humidified CO2 incubator at 37 °C.
2. Reagents. The collection of CDK inhibitors was purchased from Sigma-Aldrich, MedChemExpress, Santa Cruz Biotechnology, Enzo Life Sciences, Tocris Bioscience, Calbiochem, Merck, or Selleck Chemicals. Tozasertib was purchased from LC Laboratories. The purity of studied compounds was >95% as determined by HPLC-MS analysis. See Supporting Information Table S1 for specific vendors for each inhibitor, exact purity, and method used for purity determination.
3. Immunoblotting. Briefly, the inhibitor-treated cells were harvested and then lysed in RIPA buffer. Proteins were separated on SDS−polyacrylamide gels and electrotransferred onto nitrocellulose membranes. After blocking, the membranes were incubated with specific primary antibodies overnight, washed, and then incubated with peroxidase-conjugated secondary antibodies. Finally, peroxidase activity was detected using Pierce ECL Western blotting substrates and a CCD camera LAS-4000 (Fujifilm). Specific antibodies were purchased from Cell Signaling (anti-FAK; anti-Aurora A, clone 1G4; anti-CDK1, clone POH1; anti-CDK2, clone 78B2), Thermo Fisher Scientific (anti-pFAK, serine 732), Santa Cruz Biotechnology (anti-βactin, clone C4; anti-Aurora B, clone E-15), or Merck Millipore (antipHistone H3, serine 10; anti-RNA polymerase II, clone ARNA3; antipRNA polymerase II, serine 2, clone 3E10; anti-pRNA polymerase II, serine 5, clone 3E8).
4. Cell Cycle Analysis. Subconfluent cells were treated with different concentrations of each test compound for 24 h. The cells were trypsinized, washed with PBS, fixed with 70% ethanol, and denatured with 2 M HCl. Following neutralization, the cells were stained with propidium iodide and analyzed by flow cytometry using a 488 nm laser (BD FACS Verse with BD FACSuite software, version 1.0.6.). Cell cycle distribution was analyzed using ModFit LT (Verity Software House, version 4.1.7).
5. Kinase Inhibition Assay. CDK/cyclin complexes were assayed as previously described.37,117−120 All kinases were tested with appropriate substrates in the presence of ATP, 0.05 μCi of [γ-33P]ATP and the test compound in a reaction buffer to a total volume of 10 μL (the concentration of DMSO in the reaction never exceeded 0.2%) (see Supporting Information Table S2 for details of individual kinase reaction conditions). The reactions were stopped by the addition of 5 μL of 3% aq H3PO4. Aliquots were spotted onto P81 phosphocellulose (Whatman), washed 3× with 0.5% aq H3PO4, and air-dried. Kinase inhibition was quantified using an FLA-7000 digital image analyzer (Fujifilm). The concentration of the test compounds required to reduce CDK activity by 50% was determined from the dose−response curves and reported as the IC50 value. The concentration of ATP used in the kinase assay was determined based on the Km value for ATP of each enzyme, which was determined for each kinase using a standard assay over an appropriate range of ATP concentrations. All assays were linear with respect to time and enzyme concentration under the conditions used. All assays were performed at least in triplicate for the indicated time using an Eppendorf ThermoMixer (350 rpm, 30 °C) in a 96-well format.
6. Cellular Thermal Shift Assay. HCT-116 cells were treated with test compounds at different concentrations for 3 h, harvested, and then lysed in RIPA buffer. The soluble fraction was separated from the cell debris by centrifugation, and protein concentration was determined. Then, the samples (50 μL) were distributed into PCR tubes, preheated (rt, 1 min), and heated at a thermal gradient for 3 min in an MJ Mini thermal cycler (Bio-Rad) followed by cooling. The appropriate thermal gradient was determined from preliminary CeTSA experiments. Then, the samples were centrifuged to remove precipitated and aggregated proteins, denatured in Laemmli sample buffer, and analyzed by immunoblotting for appropriate proteins.